- Today
- Total

비둘기 둥지
[생물정보학 / python] 1.biopython - Sequence 객체 조작 본문
1. Seq함수로 Sequence 객체 만들기
## 필요 패키지 import
from Bio.Seq import Seq
## Biopython은 DNA 염기 서열을 Seq 자료형으로 사용한다.
test_seq = Seq('AGTACATGGT')
print(type(test_seq))
## Sequence의 서열 정보를 추가하도록 해줌.
#! Biopython 1.78 이후로 Alphabet이 사라졌다.. 코쓱머쓱
# from Bio import Alphabet
import Bio
Bio.__version__
## 출력 결과
<class 'Bio.Seq.Seq'>
## 작성한 코드는 biopython 1.79 버전을 사용하였다.
'1.79'2. SeqIO 여러 파일 파싱하기
2-1. FASTA 파일 파싱하기
- FASTA 파일은 텍스트 파일로 염기서열이나 단백질 서열 정보를 담고 있다.
- 첫 번쨰 줄은 > 문자로 시작하는 헤더가 있다.
- 두 번째 줄 부터는 120 글자 이하의 서열이 한 줄씩 표현되어 있다.
- Biopython은 단 한 줄로 서열을 읽어 헤더정보와 서열 정보를 구분할 수 있다.

## FASTA 파일 파싱하기
from Bio import SeqIO
for seq_record in SeqIO.parse("../dataset/fasta/ls_orchid.fasta", "fasta"):
print(seq_record.id)
print(repr(seq_record.seq), '\n')
## 출력 결과
gi|2765658|emb|Z78533.1|CIZ78533
Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC')
gi|2765657|emb|Z78532.1|CCZ78532
Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAG...GGC')
gi|2765656|emb|Z78531.1|CFZ78531
Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAGCAG...TAA')
... 중략2-2. GenBank 파일 파싱하기
- NCBI 데이터에서 대중에게 제공하는 포맷으로 염기서열과 CDS(Coding Sequence) 별로 번역된
아미노산 서열, 종의 정보, 관련 논문 저자, 제목 pubmed ID 등의 메타 데이터를 담고 있다.- 1개 이상의 GenBank 정보가 있을 수 있으며, 끝맺음 구분은 //로 한다.
- 메타 데이터가 담고 있는 정보
| 항목 | 설명 |
| LOCUS | Accession ID, 길이, 분자 종류, GenBank Division 정보, 최종 수정 날짜 |
| DEFINITION | 서열에 대한 간략한 설명 |
| ACCESSION | 서열의 독자적인 ID |
| VERSION | 서열에 변화가 생기면 버전이 올라간다. e. g.) KT225467.1 => KT225467.2 |
| KEYWORDS | - 서열을 설명하는 키워드 - NCBI에 검색할때 Keyword 항목으로 참조하는 부분 - 만약 존재하지 않는 경우에는 .으로 표기한다. |
| SOURCE | 서열의 근원에 대한 정보 |
| REFERENCE | - 서열에 관한 논문 정보가 담긴 부분 - 논문 저자, 제목, 저널명, pubmed id가 포함됨 |
| COMMENT | 기타 설명이 필요한 경우에 입력한다 |
| FEATURES | - 서열 특징이 포함되어 있는 부분 - 전체 서열 구간 정보(source)와 각 CDS 정보가 나열되어 있다. |
| ORIGIN | - 염기 서열이 표시되어 있는 부분 - 60개 염기서열을 한 줄로 하며 10개 단위로 끊어 소문자로 표기 |

## GenBank 파일 파싱하기
for gen_record in SeqIO.parse('../dataset/genbank/ls_orchid.gbk', 'genbank'):
print(gen_record.id)
print(repr(gen_record.seq), '\n')
## 출력 결과
Z78533.1
Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGATGAGACCGTGG...CGC')
Z78532.1
Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAACAG...GGC')
Z78531.1
Seq('CGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTGTTGAGACAGCAG...TAA')
3. Sequence 파일 조작하기
- biopython의 Sequence 객체는 Python의 String객체와 비슷한 특징을 가진다.
| 메소드 | 사용법 | 설명 |
| count() | sequence.count(str) | sequence에 str이 들어가 있는 갯수를 반환 |
| lower() | sequence.lower() | sequence의 문자열을 소문자로 변환 |
| upper() | sequence.upper() | sequence의 문자열을 대문자로 변환 |
| split() | sequence.split(str) | sequence을 str을 기준으로 하여 분할 |
| strip() | sequence.strip() | sequence 양끝의 공백, 엔터와 같은 문자를 제거 |
| startswith() | sequence.startswith(str) | sequence가 str로 시작하는지 판별 |
| transcribe() | sequence.transcribe() | DNA서열을 RNA로 전사한 서열을 반환 |
| translate() | sequence.translate() | DNA 또는 RNA서열을 단백질 서열로 반환 |
| complement() | sequence.complement() | sequence가 가진 서열의 상보적 서열을 반환 |
| reverse_complement() | sequence.reverse_complement() | sequence가 가진 서열의 역상보적 서열을 반환 |
## Sequence 객체는 파이썬의 string처럼 indexing과 slicing, count가 가능하다.
print(f'첫 글자 인덱싱 : {genbank_seq[0]}')
print(f'마지막 글자 인덱싱 : {genbank_seq[-1]}')
print(f'슬라이싱 : {genbank_seq[4: 15]} \n')
## Sequence 객체 더하기
sequence1 = Seq("ACGT")
sequence2 = Seq("AACC")
print(f'sequence 2개 더하기 : {sequence1 + sequence2}\n')
## 출력 결과
첫 글자 인덱싱 : C
마지막 글자 인덱싱 : C
슬라이싱 : GTTGAGATCAC
sequence 2개 더하기 : ACGTAACC## Sequence 객체 대소문자 변환
lower_seq = genbank_seq.lower()
upper_seq = lower_seq.upper()
print(f'원본 sequence \n {genbank_seq}\n')
print(f'소문자 sequence \n {lower_seq}\n')
print(f'대문자 sequence \n {upper_seq}')
## 원래의 Sequence 객체는 불변이지만, tomutable()을 통해 변하게 할 수 있다.
try:
coding_dna[5] = 'C'
except TypeError as te:
print(f'TypeError : {te}')
mutable_coding_dna = coding_dna.tomutable()
mutable_coding_dna[5] = 'G'
print(f'원본 dna : {coding_dna} \n변경된 dna : {mutable_coding_dna}')
## mutable이 된 Sequence는 toseq()로 다시 immutable로 변경할 수 있다.
immutable_coding_dna = mutable_coding_dna.toseq()
try:
coding_dna[5] = 'C'
except TypeError as te:
print(f'TypeError : {te}')
## 출력 결과
원본 sequence
CATTGTTGAGATCACATAATAATTGATCGAGTTAATCT...(중략)
소문자 sequence
cattgttgagatcacataataattgatcgagttaatct...(중략)
대문자 sequence
CATTGTTGAGATCACATAATAATTGATCGAGTTAATCT...(중략)
TypeError: 'Seq' object does not support item assignment
원본 dna : ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG
변경된 dna : ATGGCGATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG
TypeError : 'Seq' object does not support item assignment(!) 염기서열의 분리 온도와 무게 계산
- DNA 이중나선에 온도를 가하면 단일 사슬로 분리된다
- Tm은 이중 나선의 절반이 단일 나선이 될 때의 온도를 나타낸다
- GC간 결합이 AT간 결합보다 결합 힘이 강하므로, GC content가 높을수록 Tm이 높아진다
from Bio.SeqUtils import MeltingTemp as mt
## DNA 이중나선 분리 온도계산
print(f'테스트 genbank sequence의 GC-content : {GC(genbank_seq)}, Tm : {mt.Tm_Wallace(genbank_seq)}')
print(f'테스트 coding dna의 GC-content : {GC(coding_dna)}, Tm : {mt.Tm_Wallace(coding_dna)} \n')
## 염기서열의 무게 계산
from Bio.SeqUtils import molecular_weight
print(f'테스트 genbank sequence의 무게 : {molecular_weight(genbank_seq)}')
print(f'테스트 coding dna의 무게 : {molecular_weight(coding_dna)}')
## 출력 결과
테스트 genbank sequence의 GC-content : 50.0, Tm : 1776.0
테스트 coding dna의 GC-content : 56.41025641025641, Tm : 122.0
테스트 genbank sequence의 무게 : 183277.6667999999
테스트 coding dna의 무게 : 12192.7563999999983-1. GC-content 구하기
- GC-content(GC 함량)은 DNA 혹은 RNA에서의 구아닌과 사이토신의 백분율이다.
- DNA에 GC 함량이 높을수록 안정적이다.
- 응용
- 분자 생물학
중합효소 연쇄 반응(PCR)에서 프라이머로 알려진 짧은 올리고 뉴클레오티드의
GC 함량은 종종 주형 DNA에 대한 융용 온도를 예측하는데 사용한다. - 계통 분류학
비진핵 생물 분류법에서 상위 계층적 분류에서 GC 비율의 사용을 권장함.
- 분자 생물학
- sequence의 GC-content는 두 가지 방법으로 구할 수 있다.
- ((sequence 안의 G의 갯수) + (sequence 안의 C의 갯수))*100 / (염기 서열의 길이)
- biopython SeqUtils의 GC 함수 사용하기
## GC contents 계산 - count 메소드 이용
content_G = genbank_seq.count("G")
content_C = genbank_seq.count("C")
print(f'G 갯수 : {content_G}, C 갯수 : {content_C} \n전체 길이 : {len(genbank_seq)}')
print(f'genbank sequence GC content : {(content_G + content_C) *100/ len(genbank_seq)}%')
## GC contents 계산 - Bio.SeqUtils.GC 함수 사용
from Bio.SeqUtils import GC
print(f'genbank sequence GC content : {GC(genbank_seq)}%')
## 출력 결과
G 갯수 : 160, C 갯수 : 136
전체 길이 : 592
genbank sequence GC content : 50.0%
genbank sequence GC content : 50.0%3-2. 염기서열의 상보, 역상보, 전사, 번역 과정 구현
- 상보성( 相補性 / complementarity)
- DNA 복제 및 전사의 기본 원리로, 자물쇠와 열쇠 원리를 따르는 두 구조 사이의 관계를 설명
- 두 DNA 가닥 또는 RNA 가닥 서열 사이에 공유되는 특성
- 상보적인 염기쌍을 통해 세포는 한 세대에서 다른 세대로 유전정보를 복사
- 염기 서열에 저장된 정보의 손상을 찾아내 복구
- 아데닌 (A)와 티민 (T) 사이에는 2중 수소결합, 구아닌 (G)와 사이토신 (C)은 3중 수소 결합한다.
(!) 전사과정에서 DNA의 아데닌은 RNA의 우라실 (U)과 결합한다.
- 전사( 傳寫 /transcription )
- DNA에 적혀 있는 유전정보를 mRNA(messenger RNA)로 옮기는 과정
- DNA의 한 쪽 가닥만을 정보로 삼아 옮겨적고 RNA가 합성된 이후 DNA는 복구된다.
- 원핵세포 : 전사된 mRNA 그대로 번역과정으로 넘어감
진핵세포 : 중간에 끼어있는 인트론을 제거하고 엑손만을 남겨야 하므로 mRNA를 가공하는 과정을 거침.
- 번역( translation )
- DNA로부터 복제된 mRNA의 염기서열을 단백질의 아미노산 배열로 고쳐 쓰는 작업
- 세포질 내의 리보솜에서 일어나며, mRNA의 정보(코돈)을 근거로 상보적으로 결합할 수 있는
tRNA(transfer RNA)가 날아오는 아미노산들을 차례차례 연결시켜 단백질을 합성.
## DNA의 전사과정 보기
coding_dna = Seq('ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG')
## 역상보 염기서열
reverse_complement = coding_dna.reverse_complement()
## conding_dna를 전사한 messenger RNA
mRNA = coding_dna.transcribe()
print(f'원본 염기서열 : {coding_dna} \n역상보 염기서열 : {reverse_complement} \n')
print(f'상보 염기서열 : {coding_dna.complement()} \nmRNA : {mRNA}')
## mRNA 번역
tRNA = mRNA.translate()
print(f'번역된 단백질 : {tRNA}')
## 출력 결과
원본 염기서열 : ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG
역상보 염기서열 : CTATCGGGCACCCTTTCAGCGGCCCATTACAATGGCCAT
#! 번역된 문자열에서 *는 단백질 번역 과정이 끝나는 종결 코돈이다.
상보 염기서열 : TACCGGTAACATTACCCGGCGACTTTCCCACGGGCTATC
mRNA : AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG
번역된 단백질 : MAIVMGR*KGAR*
(!) Python String형 객체의 전사, 번역
- python String형 객체를 이용한 전사, 번역 과정을 구현하기 위해서는
biopython 패키지의 함수들을 이용해야한다.
## 정확히 어떤 염기인지 모를떄 UnkownSeq를 이용해 ?로 채움.
from Bio.Seq import UnknownSeq
unk_dna = UnknownSeq(20)
print(f'무슨 dna지? : {unk_dna} \n')
## Sequence 객체 파이썬 String으로 변환
string_seq = str(genbank_seq)
print(f'원본 sequence 타입 : {type(genbank_seq)} \nstring sequence 타입 : {type(string_seq)}\n')
## python string 형으로 전사, 번역 과정 시행
from Bio.Seq import reverse_complement, transcribe, translate
test_string = "GCTGTTATGGGTCGTTGGAAGGGTGGTCGTGCTGCTGGTTAG"
recv_comp = reverse_complement(test_string)
mRNA = transcribe(test_string)
translate_mRNA = translate(mRNA)
print(f'DNA string : {test_string} \n역상보 string : {recv_comp} \n')
print(f'mRNA string : {mRNA} \n번역된 mRNA : {translate_mRNA}')
## 출력 결과
원본 sequence 타입 : <class 'Bio.Seq.Seq'>
string sequence 타입 : <class 'str'>
무슨 dna지? : ????????????????????
DNA string : GCTGTTATGGGTCGTTGGAAGGGTGGTCGTGCTGCTGGTTAG
역상보 string : CTAACCAGCAGCACGACCACCCTTCCAACGACCCATAACAGC
mRNA string : GCUGUUAUGGGUCGUUGGAAGGGUGGUCGUGCUGCUGGUUAG
번역된 mRNA : AVMGRWKGGRAAG*3-3. 코돈 테이블
- 코돈 (Codon)
- 번역 과정에서 mRNA의 염기서열 3개가 하나로 묶여 한 개의 아미노산을 구성한다.
- 이 3개 염기의 조합을 트리플렛 코드라 부르며, 코돈 이라는 단위로 나타낸다.
- 단백질 합성 (번역 개시) 신호를 의미하는 개시 코돈 (Start Codon) : AUG
번역 종료 신호를 의미하는 종료 코돈 (Stop Codon) : UAA, UGA, UAG
(!) 유기체에 따라 시작 코돈으로 GUG 또는 UUG가 포함된다.
- 코돈 테이블
- 3개 염기로 생성될 수 있는 조합들을 모아둔 표.
- 유전자 코드를 아미노산 서열로 번역하는데 사용
## 코돈 테이블
## mRNA는 번역 과정에서 코돈테이블에 맞는 아미노산으로 번역된다.
from Bio.Data import CodonTable
## 표준 코돈 테이블
standard_table = CodonTable.unambiguous_dna_by_name['Standard']
print(standard_table, '\n')
## Vertebrate Mitochondrial 코돈 테이블
mito_table = CodonTable.unambiguous_dna_by_name['Vertebrate Mitochondrial']
print(mito_table)
## 출력 결과
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA Stop| A
T | TTG L(s)| TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L(s)| CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I | ACT T | AAT N | AGT S | T
A | ATC I | ACC T | AAC N | AGC S | C
A | ATA I | ACA T | AAA K | AGA R | A
A | ATG M(s)| ACG T | AAG K | AGG R | G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V | GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--
Table 2 Vertebrate Mitochondrial, SGC1
| T | C | A | G |
--+---------+---------+---------+---------+--
T | TTT F | TCT S | TAT Y | TGT C | T
T | TTC F | TCC S | TAC Y | TGC C | C
T | TTA L | TCA S | TAA Stop| TGA W | A
T | TTG L | TCG S | TAG Stop| TGG W | G
--+---------+---------+---------+---------+--
C | CTT L | CCT P | CAT H | CGT R | T
C | CTC L | CCC P | CAC H | CGC R | C
C | CTA L | CCA P | CAA Q | CGA R | A
C | CTG L | CCG P | CAG Q | CGG R | G
--+---------+---------+---------+---------+--
A | ATT I(s)| ACT T | AAT N | AGT S | T
A | ATC I(s)| ACC T | AAC N | AGC S | C
A | ATA M(s)| ACA T | AAA K | AGA Stop| A
A | ATG M(s)| ACG T | AAG K | AGG Stop| G
--+---------+---------+---------+---------+--
G | GTT V | GCT A | GAT D | GGT G | T
G | GTC V | GCC A | GAC D | GGC G | C
G | GTA V | GCA A | GAA E | GGA G | A
G | GTG V(s)| GCG A | GAG E | GGG G | G
--+---------+---------+---------+---------+--## Vertebrate Mitochondrial의 개시 코돈
init_codon = mito_table.start_codons
## Vertebrate Mitochondrial의 종결 코돈
stop_codon = mito_table.stop_codons
print(f'개시 코돈 : {init_codon} \n종결 코돈 : {stop_codon}')
## 출력 결과
개시 코돈 : ['ATT', 'ATC', 'ATA', 'ATG', 'GTG']
종결 코돈 : ['TAA', 'TAG', 'AGA', 'AGG']3-3. ORF ( Open Record Frame )
- 개시 코돈에서부터 종결코돈까지 3배수로 구성된 염기서열
- ORF들에서 실제로 단백질로 번역되는 ORF를 CDS (Coding Sequence)라고 한다.
tata_seq = Seq("TATAAAGGCAATATGCAGTAGGCAAAGGCAACGGAAGGCCGGAAAAAGGCCATGCCCGGT
GGGTTTTCCCCAGCGTGACCCGGAAAACCTGAGGAACCC")
## 시작 코돈의 위치
start_idx = tata_seq.find('ATG')
## 시작 코돈 이후 종결 코돈의 위치
end_idx = tata_seq.find('TAG', start_idx)
## open record frame
## 종결 코돈이 시작하는 인덱스 ~ 3칸 뒤의 염기 서열까지
orf = tata_seq[start_idx: end_idx + 3]
print(f'테스트 amino acid sequence의 ORF : {orf}')
print(f'ORF의 무게 : {molecular_weight(orf)}')
## 출력 결과
테스트 amino acid sequence의 ORF : ATGCAGTAG
ORF의 무게 : 2842.8206999999993(!) Six-frame translations
- DNA 이중나선은 두 가닥의 서열이 서로 상보적으로 연결되어 있고, 염기서열 3개씩 읽어 전사
- 두 가닥의 서열은 서로 구분되는 6개의 ORF를 가진다.
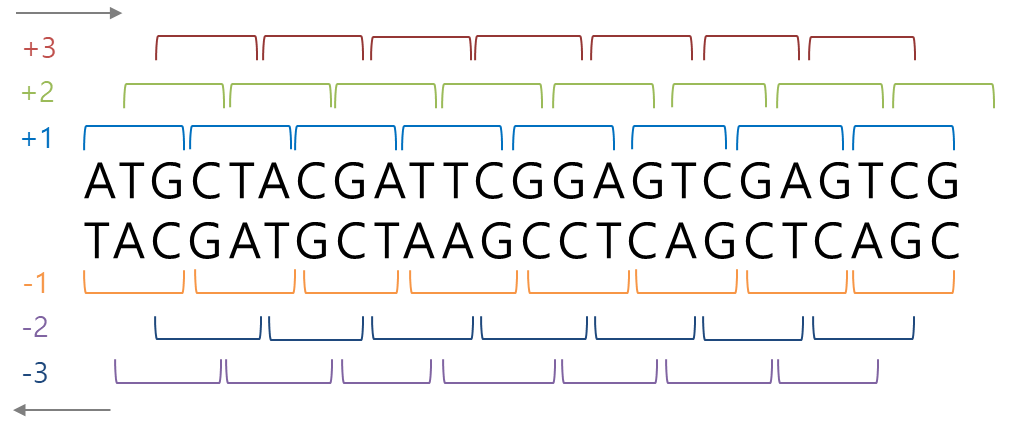
## DNA 서열에서 가능한 모든 6개의 번역된 서열 구하기
from Bio.SeqUtils import six_frame_translations
dummy_seq = Seq('ATGCCTTGAAATGTATAG')
print(f'dummy sequence six frame translation\n{"-"*58}\n{six_frame_translations(dummy_seq)}\n\n')
print(f'tata sequence six frame translation\n{"-"*58}\n{six_frame_translations(tata_seq)}\n\n')
## 출력 결과
dummy sequence six frame translation
----------------------------------------------------------
GC_Frame: a:6 t:6 g:4 c:2
Sequence: atgccttgaaatgtatag, 18 nt, 33.33 %GC
1/1
A L K C I
C L E M Y
M P * N V *
atgccttgaaatgtatag 33 %
tacggaactttacatatc
G Q F T Y
H R S I Y L
A K F H I
tata sequence six frame translation
----------------------------------------------------------
GC_Frame: a:31 t:13 g:30 c:25
Sequence: tataaaggca ... tgaggaaccc, 99 nt, 55.56 %GC
1/1
* R Q Y A V G K G N G R P E K G H A R W
I K A I C S R Q R Q R K A G K R P C P V
Y K G N M Q * A K A T E G R K K A M P G
tataaaggcaatatgcagtaggcaaaggcaacggaaggccggaaaaaggccatgcccggt 51 %
atatttccgttatacgtcatccgtttccgttgccttccggcctttttccggtacgggcca
L P L I C Y A F A V S P R F F A M G P P
I F A I H L L C L C R F A P F L G H G T
Y L C Y A T P L P L P L G S F P W A R H
61/21
V F P S V T R K T * G T
G F P Q R D P E N L R N
G F S P A * P G K P E E P
gggttttccccagcgtgacccggaaaacctgaggaaccc 61 %
cccaaaaggggtcgcactgggccttttggactccttggg
N E G A H G P F G S S G
P K G W R S G S F R L F G
T K G L T V R F V Q P V3-4. 아미노산 서열의 약자와 기호간 변환
- 아미노산 서열을 표현하는 방법은 다음과 같다.
| 아미노산 | 영어 이름 | 기호 | 약자 |
| 알라닌 | Alanine | Ala | A |
| 시스테인 | Cysteine | Cys | C |
| 아스파르트산 | Aspartic acid | Asp | D |
| 글루탐산 | Glutamic acid | Glu | E |
| 페닐알라닌 | Phenylalanine | Phe | F |
| 글라이신 | Glycine | Gly | G |
| 히스티딘 | Histidine | His | H |
| 아이소류신 | Isoleucine | Ile | I |
| 라이신 | Lysine | Lys | K |
| 류신 | Leucine | Leu | L |
| 메티오닌 | Methionine | Met | M |
| 아스파라긴 | Asparagine | Asn | N |
| 피롤라이신 | Pyrrolysine | Ply | O |
| 프롤린 | Proline | Pro | P |
| 글루타민 | Glutamine | Gln | Q |
| 아르기닌 | Arginine | Arg | R |
| 세린 | Serine | Ser | S |
| 트레오닌 | Threonine | Thr | T |
| 셀레노시스테인 | Selenocysteine | Sec | U |
| 발린 | Valin | Val | V |
| 트립토판 | Tryptophan | Trp | W |
| 타이로신 | Tyrosine | Tyr | Y |
## 아미노산 서열을 약자와 기호간 변환하기
from Bio.SeqUtils import seq1, seq3
## DNA의 번역, 전사 과정
comp_tata_seq = tata_seq.complement()
mRNA = comp_tata_seq.transcribe()
tRNA = mRNA.translate(to_stop = True)
## 약자 => 기호
protein_3 = seq3(tRNA)
## 기호 => 약자
protein_1 = seq1(protein_3)
print(f'원본 아미노산 고리 : {tRNA}')
print(f'기호 아미노산 고리 : {protein_3}, {len(protein_3)}, {len(protein_3) // 3}')
print(f'약자 아미노산 고리 : {protein_1}, {len(protein_1)}')
## 출력 결과
원본 아미노산 고리 : IFPLYVIRFRCLPAFFRYGPPKRGRTGPFGLLG
기호 아미노산 고리 : IlePheProLeuTyrValIleArgPheArgCysLeuProAlaPhePheArgTyrGlyProProLysArgGlyArgThrGlyProPheGlyLeuLeuGly, 99, 33
약자 아미노산 고리 : IFPLYVIRFRCLPAFFRYGPPKRGRTGPFGLLG, 3399. 참고 자료
99-1. 도서
- 비제이퍼블릭 | 한주현 저 - 바이오파이썬으로 만나는 생물정보학
99-2. 블로그
- 생물정보학자의 블로그 | 생물정보학/Tools - ORF와 CDS의 차이점과 refGene.txt.gz 파일
- wikipedia | GC 함량 / 상보성(분자생물학) / 전사(생물학) / 번역(생물학)
- incodom | Codon
[생물정보학] ORF와 CDS의 차이점과 refGene.txt.gz 파일
안녕하세요 한주현입니다 오늘은 ORF (Open Reading Frame)와 CDS (Coding Sequence) 의 차이점과 refGene.txt.gz 파일에 대해서 알아보겠습니다. ORF ORF (Open Reading Frame)는 시작 코돈 (AUG)에서부터 종결..
korbillgates.tistory.com
위키백과, 우리 모두의 백과사전
위키백과:대문 위키백과, 우리 모두의 백과사전. 위키백과 우리 모두가 만들어가는 자유 백과사전문서 584,903개와 최근 기여자 2,135명 사랑방 다른 분들과 의견을 교환해봐요! 질문방 지침으로
ko.wikipedia.org
인코덤-생물정보 공유의 장 - 인코덤, 생물정보 전문위키
人CoDOM은 위키(Wiki)기반의 커뮤니티 형성을 통한 생물정보 분야의 집단 지성 창출을 목적으로 운영되는 지식 커뮤니티로 생물정보 지식의 관리와 공유를 통한 범국가적 네트워크 형성 및 공유와
www.incodom.kr
전체코드
GitHub - EvoDmiK/TIL: Today I Learn
Today I Learn. Contribute to EvoDmiK/TIL development by creating an account on GitHub.
github.com
부탁 말씀
개인적으로 공부하는 과정에서 오류가 있을 수 있으니, 오류가 있는 부분은 댓글로 정정 부탁드립니다.
'생물 정보학 > python' 카테고리의 다른 글
| [생물정보학 / python] 3-1. 지금까지 공부한 내용 연습 (0) | 2022.04.16 |
|---|---|
| [생물정보학 / python] 3.biopython - Multiple Sequence Alignment (0) | 2022.04.14 |
| [생물정보학 / python] 2.biopython - SeqRecord 객체, 다양한 데이터 포맷 조작 (0) | 2022.04.10 |


