250x250
Recent Posts
Recent Comments
Archives
- Today
- Total

KimDove
안녕하세요, 딥러닝 엔지니어 김둘기 입니다.
비둘기 둥지
[생물정보학 / python] 3.biopython - Multiple Sequence Alignment 본문
728x90
(!) sequence alignment (서열 정렬)
(1) 유전자나 단백질의 서열을 다이내믹 프로그래밍 기반의 컴퓨터 스트링 정렬 알고리즘을 이용하여 배열
(2) 쌍서열정렬(pairwise sequence alignment)와 다중서열정렬(multiple sequence alignment)로 나뉜다.1. Multiple Sequence Alignment (MSA / 다중 서열 정렬)
- 3개 이상의 DNA, RNA, Protein과 같은 서열들을 sequence alignment를 진행하는 것을 말한다.
- 단백질 서열간 유사성을 통해 단백질의 기능을 이해, 구조 예측, 진화 관계 규명하는데 사용된다.
- Multiple Sequence Alignment를 하기 위해서는 3가지 단계가 필요하다.
- 한 서열과 비교할 여러 서열이 필요하다
- MUSCLE 툴을 통해 Multiple Sequence Alignment를 수행한다.
- Multiple Sequence Aligment를 시각화 한다.
1-1. biopython에서의 Multiple Sequence Alignment
- biopython에서 Multiple Sequence Alignment 파일을 읽기 위해 AlignIO 모듈의 read메소드를 사용한다.
- Align.read() 메소드는 두 입력 인자를 받는다.
- 파일이나 파일을 연 객체
- multiple sequence가 담긴 파일 종류
a. Alignment 다루기
- AlignIO 파일을 읽어들여 생성된 MultipleSeqAlignment 객체에 SeqRecord 객체가 들어있다.
- SeqRecord 객체의 seq, id 속성은 파이썬의 문자열과 같이 슬라이싱이 가능하다
from Bio import AlignIO
## multi alignment 파일 읽기
alignment = AlignIO.read('../dataset/aln/example.aln', "clustal")
print(f'alignment : {type(alignment)}, {len(alignment)}\n{alignment}\n')
for idx, record in enumerate(alignment, 1):
print(f'[{idx}] Alignment 레코드 정보 ({type(record)})\nid : {record.id} \nSequence : {record.seq[:70]} \n')
## 출력 결과
alignment : <class 'Bio.Align.MultipleSeqAlignment'>, 3
Alignment with 3 rows and 142 columns
MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYF...KYR sp|P69905|HBA_HUMAN
MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYF...KYR sp|P69907|HBA_PANTR
MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYF...KYR sp|P01942|HBA_MOUSE
[1] Alignment 레코드 정보 (<class 'Bio.SeqRecord.SeqRecord'>)
id : sp|P69905|HBA_HUMAN
Sequence : MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNA
[2] Alignment 레코드 정보 (<class 'Bio.SeqRecord.SeqRecord'>)
id : sp|P69907|HBA_PANTR
Sequence : MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNA
[3] Alignment 레코드 정보 (<class 'Bio.SeqRecord.SeqRecord'>)
id : sp|P01942|HBA_MOUSE
Sequence : MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHGKKVADALASAb. MUSCLE (Multiple Sequence Comparison by Log-Expectation)
- 대표적인 alignment tool로 1초에 수백기의 서열을 alignment할 수 있음.
- Phylogenetic tree 구성을 위한 대량의 데이터를 활용하기에 매우 유용함.
- 기존 Alignment 정보에 새로운 서열을 추가하여 update하거나 각 구성되어 있는 alignment profile간 비교도 가능
- 설치 및 사용법
-> 설치 링크에서 파일을 다운로드 받고 압축을 해제한 후에 터미널에 다음 커맨드를 입력한다.
| OS | 설치 링크 | 실행법 |
| Linux | http://www.drive5.com/muscle/downloads3.8.31/muscle3.8.31_i86linux64.tar.gz | muscle3.8.31_i86linux64 -in in.fasta -out out.aln |
| Mac OSX | http://www.drive5.com/muscle/downloads3.8.31/muscle3.8.31_i86darwin64.tar.gz | muscle3.8.31_i86darwin64 -in in.fasta -out out.aln |
| Windows | http://www.drive5.com/muscle/downloads3.8.31/muscle3.8.31_i86win32.exe | muscle3.8.31_i86win32 -in in.fasta -out out.aln |
b-(1). 바이오파이썬을 통한 MUSCLE 실행
- 바이오파이썬에서 MUSCLE을 실행하기 위해서는 MUSCLE이 다운로드되어 있어야 한다.
- MuscleCommandline함수에서 MUSCLE의 실행 파일 경로를 인자값으로 받는다.
from Bio.Align.Applications import MuscleCommandline
import os
muscle_path = '../utils/muscle'
cmd_line = MuscleCommandline(muscle_path, input = '../dataset/fasta/HBA.all.fasta',
out="../dataset/aln/HBA.all.aln", clw=" ")
## cmd_line에 저장되어 있는 명령어가 터미널에서 입력되는것과 같은 역할을 함.
## 이 코드와 비슷한 역할
# os.system('../utils/muscle -in ../dataset/fasta/HBA.all.fasta
# -out ../dataset/aln/HBA-2.all.aln -clw')
print(f'command line : {cmd_line}')
std_out, std_err = cmd_line()
## 출력 결과
command line : ../utils/muscle -in ../dataset/fasta/HBA.all.fasta -out ../dataset/aln/HBA.all.aln -clw
(!) biopython에서 MUSCLE을 수행한 결과
MUSCLE (3.8) multiple sequence alignment
sp|P01994|HBA_CHICK MVLSAADKNNVKGIFTKIAGHAEEYGAETLERMFTTYPPTKTYFPHFDLSHGSAQIKGHG
sp|P18971|HBA_BALAC MVLSPTDKSNVKATWAKIGNHGAEYGAEALERMFMNFPSTKTYFPHFDLGHDSAQVKGHG
sp|P01948|HBA_RABIT MVLSPADKTNIKTAWEKIGSHGGEYGAEAVERMFLGFPTTKTYFPHFDFTHGSEQIKAHG
sp|P01942|HBA_MOUSE MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHG
sp|P69907|HBA_PANTR MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHG
sp|P69905|HBA_HUMAN MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHG
sp|P01958|HBA_HORSE MVLSAADKTNVKAAWSKVGGHAGEYGAEALERMFLGFPTTKTYFPHFDLSHGSAQVKAHG
**** **.*:* : *:. *. *****::**** :*.*********. *.* *:*.**
sp|P01994|HBA_CHICK KKVVAALIEAANHIDDIAGTLSKLSDLHAHKLRVDPVNFKLLGQCFLVVVAIHHPAALTP
sp|P18971|HBA_BALAC KKVADALTKAVGHMDNLLDALSDLSDLHAHKLRVDPANFKLLSHCLLVTLALHLPAEFTP
sp|P01948|HBA_RABIT KKVSEALTKAVGHLDDLPGALSTLSDLHAHKLRVDPVNFKLLSHCLLVTLANHHPSEFTP
sp|P01942|HBA_MOUSE KKVADALASAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLASHHPADFTP
sp|P69907|HBA_PANTR KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTP
sp|P69905|HBA_HUMAN KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTP
sp|P01958|HBA_HORSE KKVGDALTLAVGHLDDLPGALSNLSDLHAHKLRVDPVNFKLLSHCLLSTLAVHLPNDFTP
*** ** *. *:*:: .:** *************.*****.:*:* .:* * * :**
sp|P01994|HBA_CHICK EVHASLDKFLCAVGTVLTAKYR
sp|P18971|HBA_BALAC SVHASLDKFLASVSTVLTSKYR
sp|P01948|HBA_RABIT AVHASLDKFLANVSTVLTSKYR
sp|P01942|HBA_MOUSE AVHASLDKFLASVSTVLTSKYR
sp|P69907|HBA_PANTR AVHASLDKFLASVSTVLTSKYR
sp|P69905|HBA_HUMAN AVHASLDKFLASVSTVLTSKYR
sp|P01958|HBA_HORSE AVHASLDKFLSSVSTVLTSKYR
*********. *.****:***2. WebLogo 표시
- Multi Sequence Alignment에서 각 행 서열의 공통부분 비율만큼 시각해 그려줌.
- WebLogo 그려주는 홈페이지 | 링크
%%time
from collections import Counter
from Bio import SeqIO
import pandas as pd
import numpy as np
sequences = SeqIO.parse('../dataset/fasta/example.fasta', 'fasta')
## 문자열 자릿수에 엄기 갯수를 구해주는 부분
def count_table(seq_list):
counter_table = {}
for idx, seq in enumerate(seq_list):
count_acid = Counter(seq)
default_dict = {}
for acid in ('A', 'G', 'C', 'T'):
default_dict[acid] = count_acid[acid] if acid in count_acid.keys() else 0
counter_table[idx] = default_dict
return counter_table
seq_list = []
for sequence in sequences: seq_list.append(sequence.seq)
print(f'염기서열 행렬 : \n{np.array(sequence_dict)}\n')
t_seq_list = np.transpose(seq_list)
print(f'염기서열 전치 행렬 : \n{t_seq_list}\n')
acid_dataframe = pd.DataFrame(count_table(t_seq_list))
print(f'각 자리별 염기서열 갯수 : \n{acid_dataframe}')
## 출력 결과
염기서열 행렬 :
[['T' 'A' 'C' 'A' 'A']
['T' 'A' 'C' 'G' 'C']
['T' 'A' 'C' 'A' 'C']
['T' 'A' 'C' 'C' 'C']
['A' 'A' 'C' 'C' 'C']
['A' 'A' 'T' 'G' 'C']
['A' 'A' 'T' 'G' 'C']]
염기서열 전치 행렬 :
[['T' 'T' 'T' 'T' 'A' 'A' 'A']
['A' 'A' 'A' 'A' 'A' 'A' 'A']
['C' 'C' 'C' 'C' 'C' 'T' 'T']
['A' 'G' 'A' 'C' 'C' 'G' 'G']
['A' 'C' 'C' 'C' 'C' 'C' 'C']]
각 자리별 염기서열 갯수 :
0 1 2 3 4
A 3 7 0 2 1
G 0 0 0 3 0
C 0 0 5 2 6
T 4 0 2 0 0
CPU times: user 10 ms, sys: 1e+03 µs, total: 11 ms
Wall time: 10.8 ms2-1. biopython으로 WebLogo 그리기
- biopython에서 WebLogo를 그리기 위한 방법으로는 두 가지 과정을 거친다.
- biopython motifs 모듈의 create() 메소드를 이용해 Motif 객체를 만듦.
- biopython Motif 모듈의 weblogo() 메소드로 weblogo를 그린다.
%time
from Bio.motifs import Motif
from Bio.Seq import Seq
from Bio import motifs
sequences = SeqIO.parse('../dataset/fasta/example.fasta', 'fasta')
instances = []
for sequence in sequences: instances.append(Seq(sequence.seq))
m = motifs.create(instances)
print(f'각 자리별 염기서열 갯수 2 : \n{m.counts}')
Motif.weblogo(m, '../Weblogo/dummy.png')
## 주피터 내에서 생성한 Weblogo 파일 보이기
from IPython.display import Image
Image('../Weblogo/dummy.png')
## 출력 결과
CPU times: user 0 ns, sys: 0 ns, total: 0 ns
Wall time: 15.5 µs
각 자리별 염기서열 갯수 2 :
0 1 2 3 4
A: 3.00 7.00 0.00 2.00 1.00
C: 0.00 0.00 5.00 2.00 6.00
G: 0.00 0.00 0.00 3.00 0.00
T: 4.00 0.00 2.00 0.00 0.00
2-2. Multi Sequence Alignment WebLogo 그리기
- biopython 1.78 이후로 Bio.alphabet 기능이 사라져 motifs.create 함수에서 alphabet을 인자값으로 직접 넣어주었다
hba_MSA = AlignIO.read('../dataset/aln/HBA.aln', 'clustal')
proteins = []
alphabet_set = set()
for hba in hba_MSA:
alphabet_set = alphabet_set.union(set(hba.seq))
proteins.append(hba.seq)
alphabet_set = ''.join(alphabet_set)
print(alphabet_set)
m = motifs.create(proteins, alphabet=alphabet_set)
Motif.weblogo(m, '../Weblogo/hba_multiple.png')
Image('../Weblogo/hba_multiple.png')
## 출력 결과
VLEPFNIASHTGKDYQRWMC
3. 계통수 그리기
- 계통수한 생물 종 간 유전적 유사도를 가지고 멀고 가까움을 그림으로 표현한 것이다.
- 계통수를 표현하는 파일 종류는 다양하며 newick 포맷을 가지고 계통수를 그린다.
- Sequence Alignment 파일에서 newick 파일로 만드는 홈페이지 | 링크
3-1. biopython으로 계통수 그리기
- Phylo.read() 메소드로 newick 파일을 읽는다.
- Phylo.draw() 메소드로 계통수 그림을 그린다.
from Bio import Phylo
tree = Phylo.read('../dataset/newick/HBA.newick', 'newick')
print(tree)
Phylo.draw(tree)
## 출력 결과
Tree(rooted=False, weight=1.0)
Clade()
Clade(branch_length=0.00537)
Clade(branch_length=0.29387, name='sp|P01994|HBA_CHICK')
Clade(branch_length=0.01203)
Clade(branch_length=0.01282)
Clade(branch_length=0.1181, name='sp|P60529|HBA_CANLF')
Clade(branch_length=0.10357, name='sp|P01948|HBA_RABIT')
Clade(branch_length=0.00617)
Clade(branch_length=0.05673)
Clade(branch_length=0.0, name='sp|P69907|HBA_PANTR')
Clade(branch_length=0.0, name='sp|P69905|HBA_HUMAN')
Clade(branch_length=0.07405, name='sp|P01958|HBA_HORSE')
Clade(branch_length=0.13313, name='sp|P18971|HBA_BALAC')
Clade(branch_length=0.06808, name='sp|P01942|HBA_MOUSE')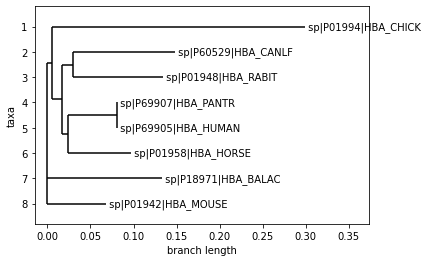
99. 자료 출처
99-1. 도서
- 비제이퍼블릭 | 한주현 저 - 바이오파이썬으로 만나는 생물정보학
99-2. 웹 사이트
99-3. 데이터셋 출처
- 바이오 파이썬으로 만나는 생물정보학 깃헙 | 깃헙 링크
전체코드
GitHub - EvoDmiK/TIL: Today I Learn
Today I Learn. Contribute to EvoDmiK/TIL development by creating an account on GitHub.
github.com
내용 추가 이력
부탁 말씀
개인적으로 공부하는 과정에서 오류가 있을 수 있으니, 오류가 있는 부분은 댓글로 정정 부탁드립니다.
728x90
'생물 정보학 > python' 카테고리의 다른 글
| [생물정보학 / python] 3-1. 지금까지 공부한 내용 연습 (0) | 2022.04.16 |
|---|---|
| [생물정보학 / python] 2.biopython - SeqRecord 객체, 다양한 데이터 포맷 조작 (0) | 2022.04.10 |
| [생물정보학 / python] 1.biopython - Sequence 객체 조작 (2) | 2022.03.30 |
'생물 정보학/python' Related Articles
more



Comments